Positive set (eQTLGen significant)
Load data
fnames = list(mixQTL = c('Whole_Blood-x-result-mixqtl-IN-eqtlgen_pos.subsample100000-with-gene-qc.txt.gz', 'gene', 'variant_id', 'pval.meta'), trcQTL = c('Whole_Blood-x-result-mixqtl-IN-eqtlgen_pos.subsample100000-with-gene-qc.txt.gz', 'gene', 'variant_id', 'pval.trc'), ascQTL = c('Whole_Blood-x-result-mixqtl-IN-eqtlgen_pos.subsample100000-with-gene-qc.txt.gz', 'gene', 'variant_id', 'pval.asc'), eQTL = c('Whole_Blood.allpairs-IN-eqtlgen_pos.subsample100000-with-gene-qc.txt.gz', 'gene_id', 'variant_id', 'pval_nominal'))
df = data.frame()
for(f in names(fnames)) {
sub = read.table(paste0(datadir, '/', fnames[[f]][1]), header = T)
if(f == 'eQTL') {
pval__ = calc_pval_from_slope(sub$slope, sub$slope_se)
sub[ ,fnames[[f]][4]] = pval__
}
sub = sub[, fnames[[f]][2:4]]
colnames(sub) = c('gene', 'variant', 'pval')
sub$method = f
sub$gene = trim_dot(sub$gene)
df = rbind(df, sub)
}
df_clean = df %>% group_by(gene, variant) %>% summarize(nobs = n()) %>% filter(nobs == 4) %>% ungroup()
df = df %>% filter(paste(gene, variant) %in% paste(df_clean$gene, df_clean$variant))
# df$pval[df$method == 'metaQTL'] = df$pval[df$method == 'metaQTL']
df %>% group_by(method) %>% summarize(nobs = n()) %>% pander
| ascQTL |
96660 |
| eQTL |
96660 |
| mixQTL |
96660 |
| trcQTL |
96660 |
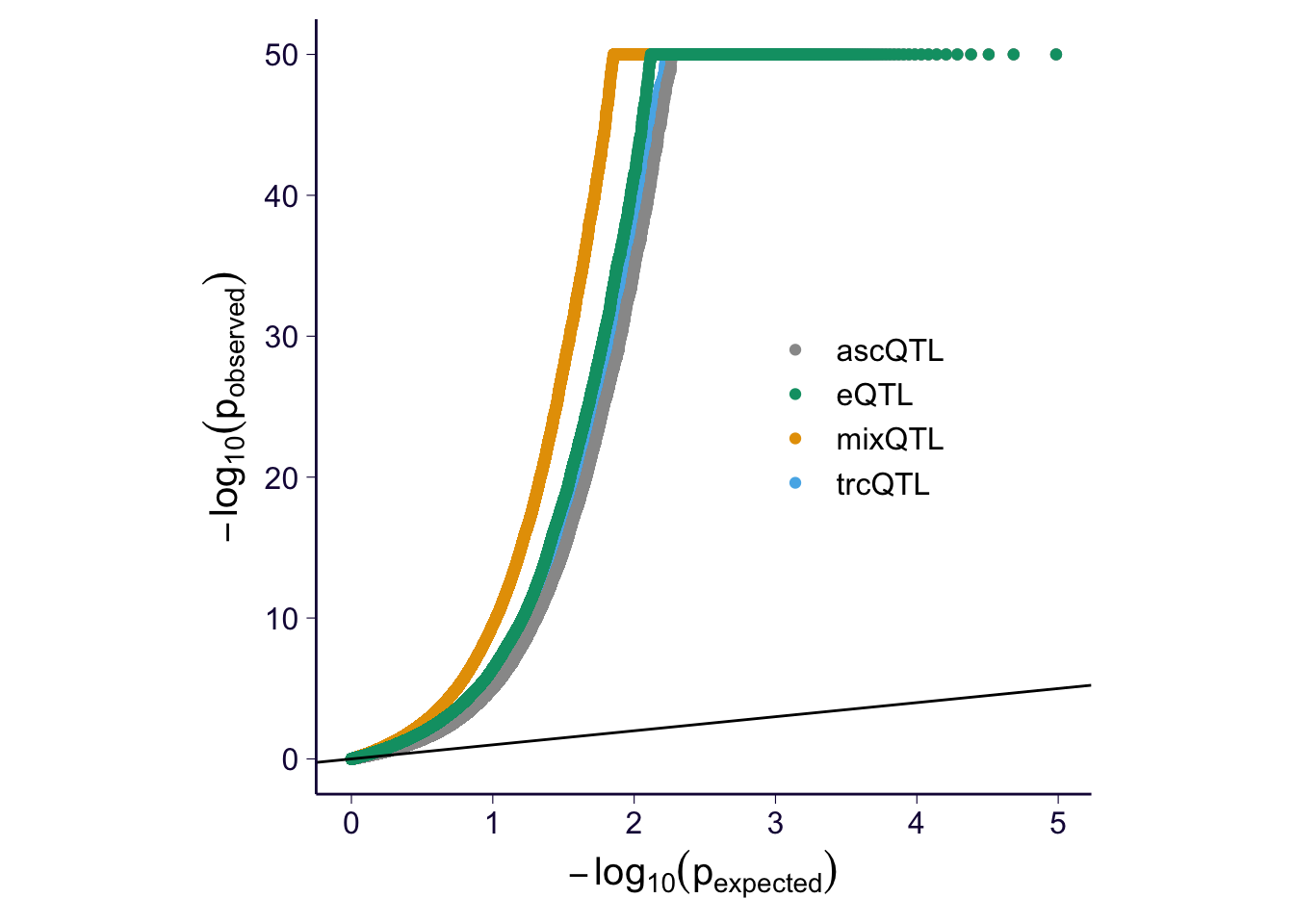
QQ-plot
df$pval[is.na(df$pval)] = runif(sum(is.na(df$pval)))
p = my_qqplot(df$pval, df$method)
p = p + geom_abline(intercept = 0, slope = 1)
p = p + scale_color_manual(values = cbPalette) + theme(legend.position = c(0.7, 0.5), legend.title = element_blank(), legend.text = element_text(size = 12)) + th + theme(aspect.ratio = 1) + ylab(expression(-log[10](p[observed]))) + xlab(expression(-log[10](p[expected])))
p$layers[[2]]$aes_params$size = 0.5
p
\(\chi^2\) plot: mixQTL vs eQTL
dfplot = df %>% mutate(chisq = qchisq(pval, df = 1, lower.tail = F)) %>% select(-pval) %>% filter(method %in% c('mixQTL', 'eQTL')) %>% dcast(gene + variant ~ method, value.var = 'chisq')
p = dfplot %>% ggplot() +
geom_point(aes(x = eQTL, y = mixQTL), alpha = 0.5) +
geom_point(data = dfplot %>% filter(gene %in% c('ENSG00000115607', 'ENSG00000213462')), aes(x = eQTL, y = mixQTL, color = gene)) +
th +
xlab(expression(paste(chi^2, '(eQTL)')))+
ylab(expression(paste(chi^2, '(mixQTL)'))) +
theme(legend.position = c(0.75, 0.15), legend.text = element_text(size = 10), legend.title = element_blank()) +
coord_equal() +
geom_abline(slope = 1, intercept = 0, color = 'lightgreen', linetype = 2, size = 1)
p
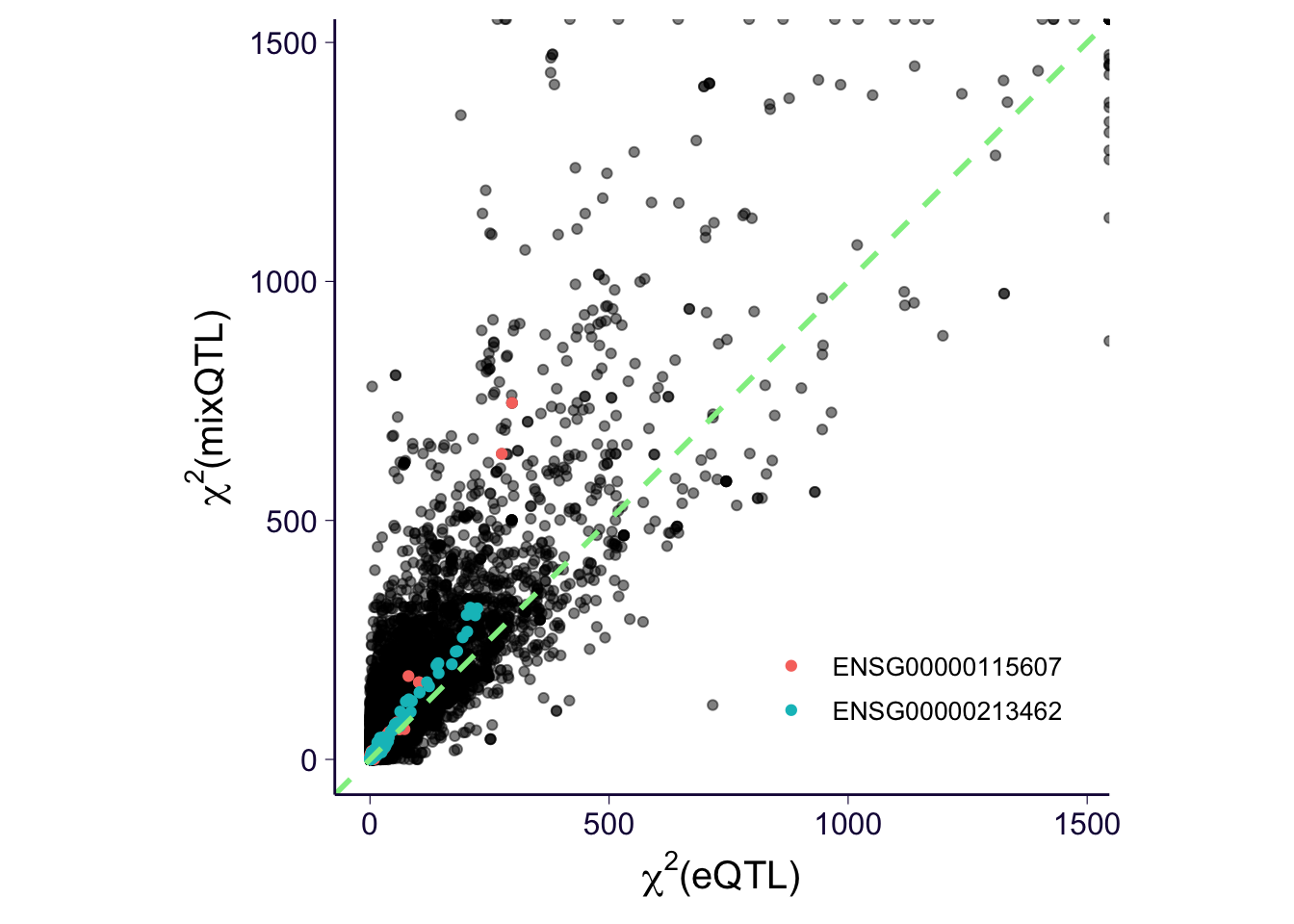
dfplot %>% summarize(median_eqtl = median(eQTL), median_mixqtl = median(mixQTL)) %>% mutate(ratio = median_mixqtl / median_eqtl) %>% pander('ratio of median')
ratio of median
| 2.588 |
3.563 |
1.377 |
dfplot %>% mutate(ratio = mixQTL / eQTL) %>% summarize(median_ratio = median(ratio, na.rm = T)) %>% pander('median of ratio')
ggsave(paste0('../output/gtex-v8-pipeline-eqtl-matrix-with-gene-qc-eqtlgen-positive-chisq.png'), p, height = 4, width = 4)
Negative set (eQTLGen pvalue > 0.5)
Load data
fnames = list(mixQTL = c('Whole_Blood-x-result-mixqtl-IN-eqtlgen_neg.subsample100000-with-gene-qc.txt.gz', 'gene', 'variant_id', 'pval.meta'), trcQTL = c('Whole_Blood-x-result-mixqtl-IN-eqtlgen_neg.subsample100000-with-gene-qc.txt.gz', 'gene', 'variant_id', 'pval.trc'), ascQTL = c('Whole_Blood-x-result-mixqtl-IN-eqtlgen_neg.subsample100000-with-gene-qc.txt.gz', 'gene', 'variant_id', 'pval.asc'), eQTL = c('Whole_Blood.allpairs-IN-eqtlgen_neg.subsample100000-with-gene-qc.txt.gz', 'gene_id', 'variant_id', 'pval_nominal'))
df = data.frame()
for(f in names(fnames)) {
sub = read.table(paste0(datadir, '/', fnames[[f]][1]), header = T)
if(f == 'eQTL') {
pval__ = calc_pval_from_slope(sub$slope, sub$slope_se)
sub[ ,fnames[[f]][4]] = pval__
}
sub = sub[, fnames[[f]][2:4]]
colnames(sub) = c('gene', 'variant', 'pval')
sub$method = f
sub$gene = trim_dot(sub$gene)
df = rbind(df, sub)
}
df_clean = df %>% group_by(gene, variant) %>% summarize(nobs = n()) %>% filter(nobs == 4) %>% ungroup()
df = df %>% filter(paste(gene, variant) %in% paste(df_clean$gene, df_clean$variant))
# df$pval[df$method == 'metaQTL'] = df$pval[df$method == 'metaQTL']
df %>% group_by(method) %>% summarize(nobs = n()) %>% pander
| ascQTL |
78691 |
| eQTL |
78691 |
| mixQTL |
78691 |
| trcQTL |
78691 |
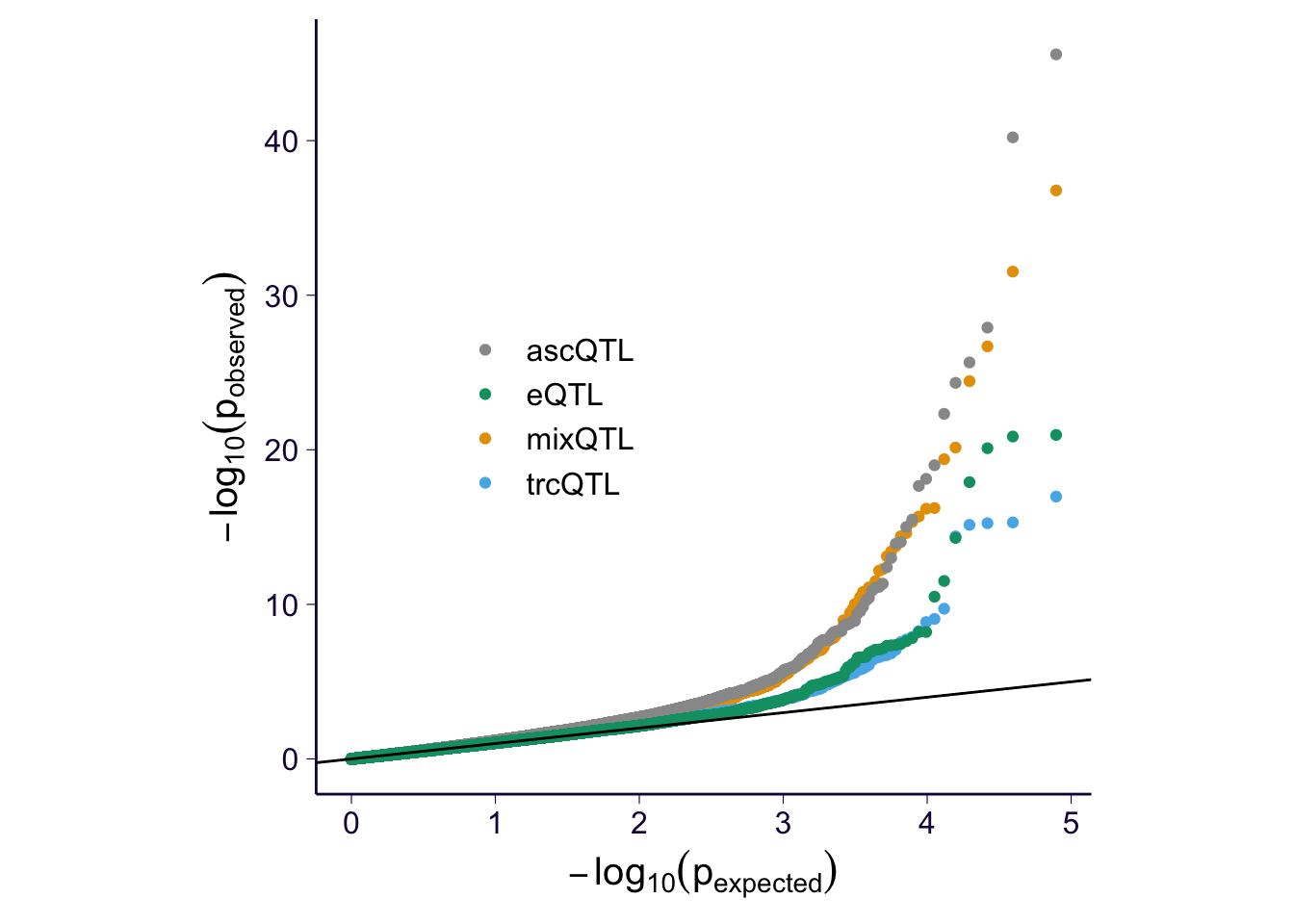
QQ-plot
df$pval[is.na(df$pval)] = runif(sum(is.na(df$pval)))
p = my_qqplot(df$pval, df$method)
p = p + geom_abline(intercept = 0, slope = 1)
p = p + scale_color_manual(values = cbPalette) + theme(legend.position = c(0.3, 0.5), legend.title = element_blank(), legend.text = element_text(size = 12)) + th + theme(aspect.ratio = 1) + ylab(expression(-log[10](p[observed]))) + xlab(expression(-log[10](p[expected])))
p$layers[[2]]$aes_params$size = 0.5
p
collector = rbind(collector %>% mutate(type = 'positive'), df %>% mutate(type = 'negative'))
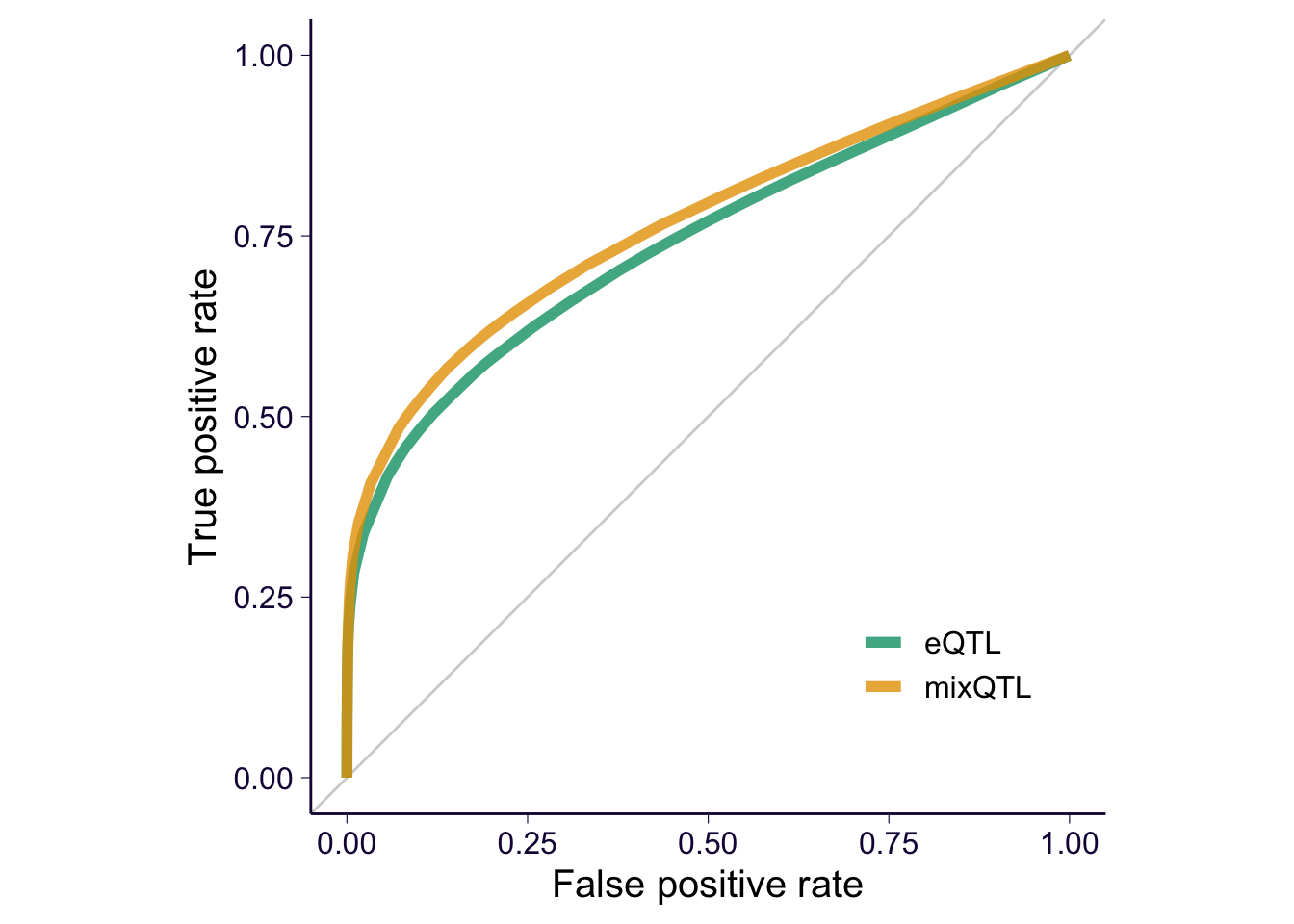
ROC/PR curves
ROC
# source('https://gist.githubusercontent.com/liangyy/6d4314dbc238236731e134abef2484f4/raw/f6b23f7f06ec97c27dff574ec1712c9d1f0552df/rlib_roc_pr.R')
collector = collector %>% mutate(eqtl = paste(gene, variant))
true_eqtls = collector %>% filter(type == 'positive')
true_eqtls = unique(true_eqtls$eqtl)
mixqtl = collector %>% filter(method == 'mixQTL')
eqtl = collector %>% filter(method == 'eQTL')
cur_mix = gen_roc_curve(true_genes = true_eqtls, gene = mixqtl$eqtl, score = -log(mixqtl$pval), method = 'gt', cutoff = c(0, 1:20/10, 10:15/5, 4:50, Inf))
cur_eqtl = gen_roc_curve(true_genes = true_eqtls, gene = eqtl$eqtl, score = -log(eqtl$pval), method = 'gt', cutoff = c(0, 1:20/10, 10:15/5, 4:50, Inf))
e2 = rbind(cur_mix %>% mutate(method = 'mixQTL'), cur_eqtl %>% mutate(method = 'eQTL'))
p = e2 %>% ggplot() + geom_abline(intercept = 0, slope = 1, color = 'lightgray') + geom_path(aes(x = fpr, y = tpr, color = method), size = 2, alpha = .8) + coord_equal()
p = p + th
p = p + xlab('False positive rate') + ylab('True positive rate') + theme(legend.position = c(0.8, 0.2), legend.title = element_blank(), legend.text = element_text(size = 12)) + scale_color_manual(values = cbPalette)
p
PR
cur_mix = gen_fdr_power_curve(true_genes = true_eqtls, gene = mixqtl$eqtl, score = -log(mixqtl$pval), method = 'gt', cutoff = c(0, 1:20/10, 10:15/5, 4:50, Inf))
cur_eqtl = gen_fdr_power_curve(true_genes = true_eqtls, gene = eqtl$eqtl, score = -log(eqtl$pval), method = 'gt', cutoff = c(0, 1:20/10, 10:15/5, 4:50, Inf))
e = rbind(cur_mix %>% mutate(method = 'mixQTL') %>% filter(recall != 0) , cur_eqtl %>% mutate(method = 'eQTL') %>% filter(recall != 0))
p2 = e %>% ggplot() + geom_path(aes(x = recall, y = precision, color = method), size = 2, alpha = .8) + theme(aspect.ratio = 1)
p2 = p2 + th
p2 = p2 + xlab('Recall') + ylab('Precision') + theme(legend.position = c(0.2, 0.2), legend.title = element_blank(), legend.text = element_text(size = 12)) + scale_color_manual(values = cbPalette)
p2
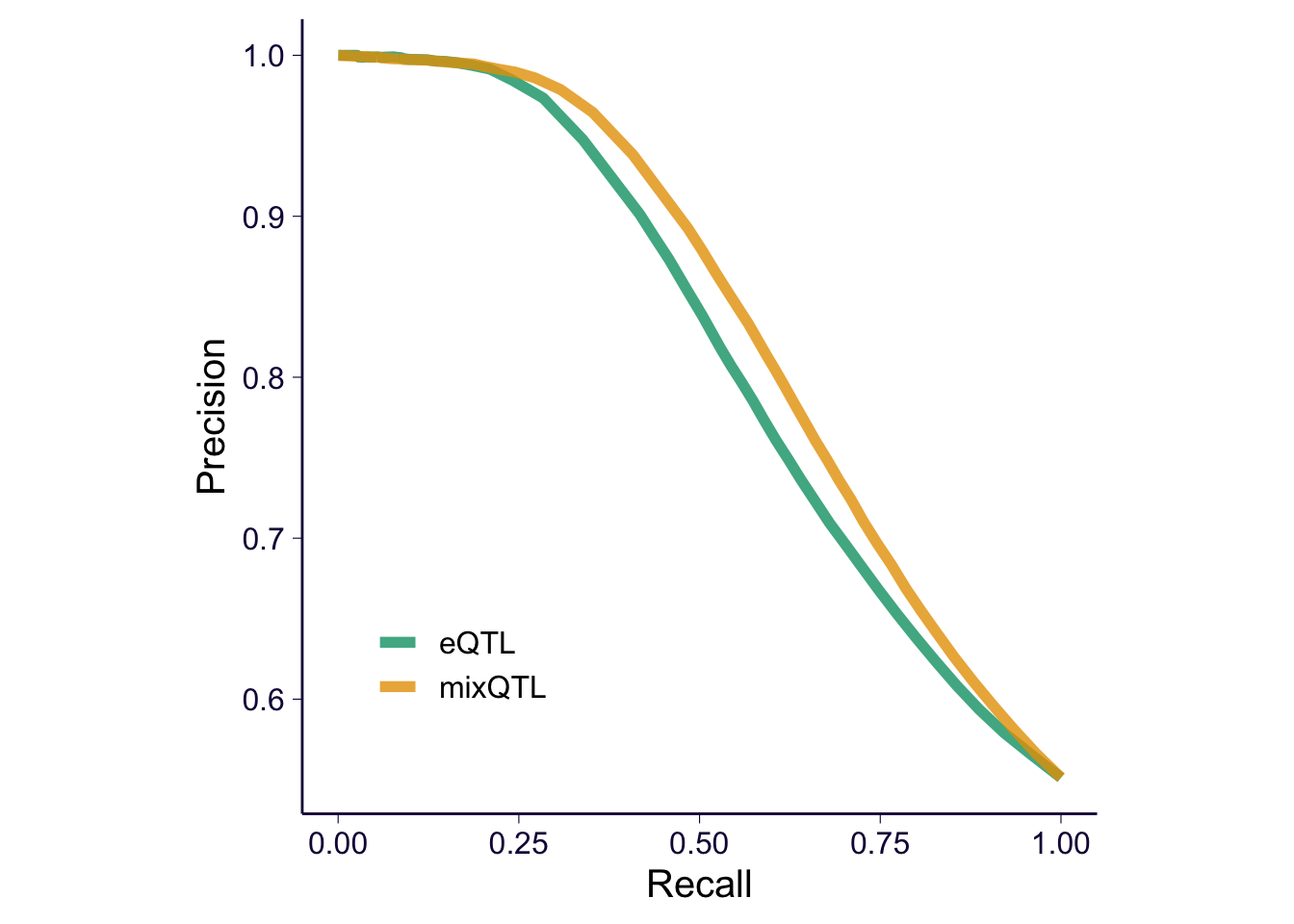
Save ROC/PR curves
ggsave('../output/gtex-v8-pipeline-eqtl-matrix-with-gene-qc-eqtlgen-roc.png', p, width = 4, height = 4)
ggsave('../output/gtex-v8-pipeline-eqtl-matrix-with-gene-qc-eqtlgen-pr.png', p2, width = 4, height = 4)
ROC/PR curves stratified by expression level
th2 = th
th2$panel.border = element_rect(colour = th2$axis.line$colour)
expression_level = read.table('/Users/yanyul/Desktop/mixqtl-pipeline-results/misc_data/gtex-v8-genes-passed-qc-with-median-trc.txt', header = T)
quantile(expression_level$median_trc, probs = 0:10/10)
## 0% 10% 20% 30% 40% 50% 60% 70% 80%
## 181.00 506.30 738.00 1018.00 1357.10 1808.50 2447.40 3456.35 5149.10
## 90% 100%
## 9985.05 5893511.00
ROC by expression level
thresholds = quantile(expression_level$median_trc, probs = c(1/3, 2/3))
collector = collector %>% mutate(expression_level = 'high')
collector$expression_level[collector$gene %in% expression_level$gene[expression_level$median_trc < thresholds[1]]] = 'low'
collector$expression_level[collector$gene %in% expression_level$gene[expression_level$median_trc < thresholds[2] & expression_level$median_trc >= thresholds[1]]] = 'middle'
roc_c = list()
for(i in unique(collector$expression_level)) {
sub = collector %>% filter(expression_level == i)
true_eqtls = sub %>% filter(type == 'positive')
true_eqtls = unique(true_eqtls$eqtl)
mixqtl = sub %>% filter(method == 'mixQTL')
eqtl = sub %>% filter(method == 'eQTL')
cur_mix = gen_roc_curve(true_genes = true_eqtls, gene = mixqtl$eqtl, score = -log(mixqtl$pval), method = 'gt', cutoff = c(0, 1:20/10, 10:15/5, 4:50, Inf))
cur_eqtl = gen_roc_curve(true_genes = true_eqtls, gene = eqtl$eqtl, score = -log(eqtl$pval), method = 'gt', cutoff = c(0, 1:20/10, 10:15/5, 4:50, Inf))
e = rbind(cur_mix %>% mutate(method = 'mixQTL'), cur_eqtl %>% mutate(method = 'eQTL'))
roc_c[[length(roc_c) + 1]] = e %>% mutate(expression_level = i)
}
roc_c = do.call(rbind, roc_c)
roc_c$expression_level = factor(roc_c$expression_level, levels = c('high', 'middle', 'low'))
p = roc_c %>% ggplot() + geom_abline(intercept = 0, slope = 1, color = 'lightgray') + geom_path(aes(x = fpr, y = tpr, color = method), size = 2, alpha = .8) + facet_wrap(~expression_level) + coord_equal() # + geom_point(aes(x = fpr, y = tpr))
p = p + th2
p = p + xlab('False positive rate') + ylab('True positive rate') + scale_color_manual(values = cbPalette) + theme(legend.position = c(0.57, 0.2), legend.title = element_blank(), legend.text = element_text(size = 12))
p
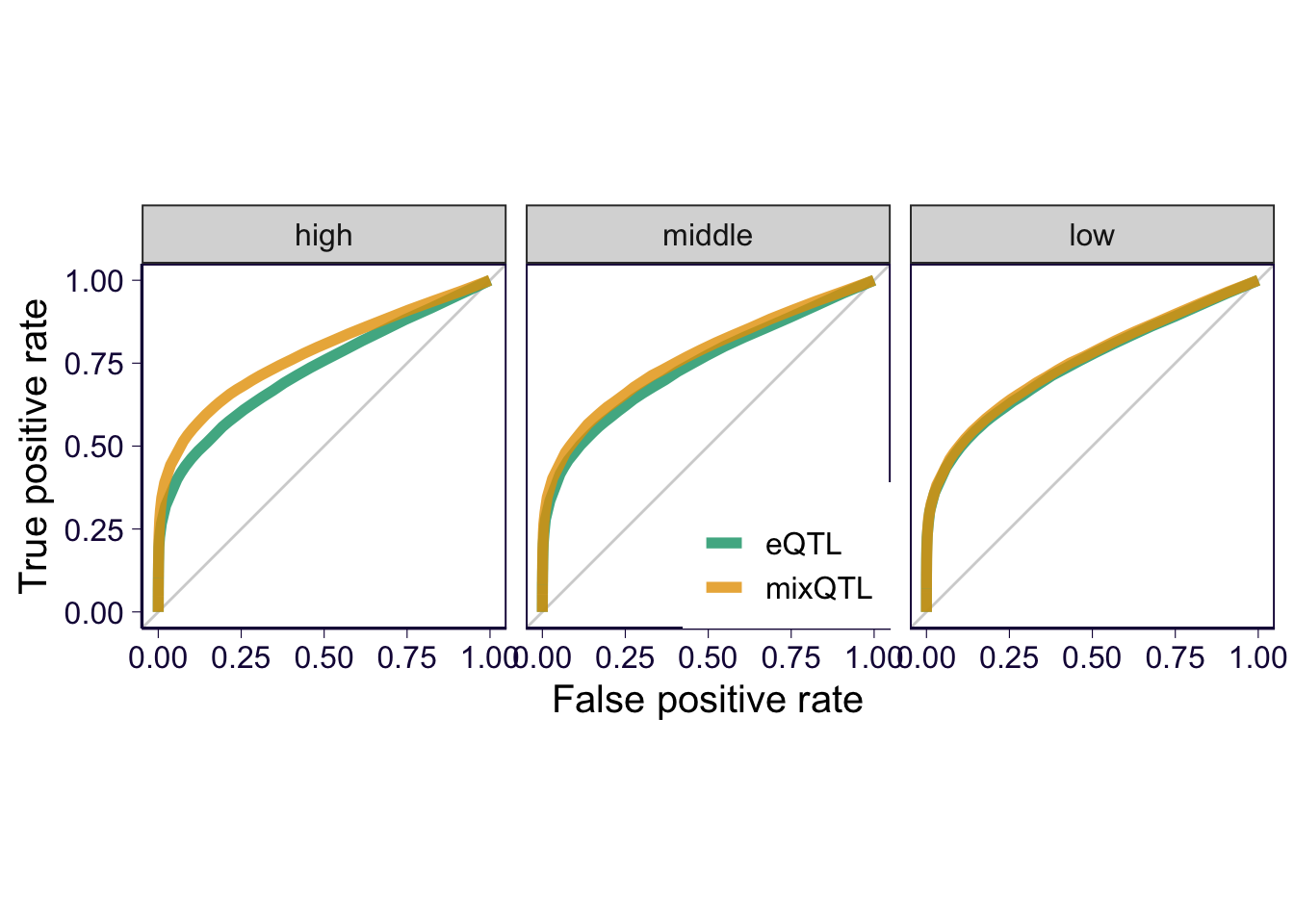
PR by expression level
pr_c = list()
for(i in unique(collector$expression_level)) {
sub = collector %>% filter(expression_level == i)
true_eqtls = sub %>% filter(type == 'positive')
true_eqtls = unique(true_eqtls$eqtl)
mixqtl = sub %>% filter(method == 'mixQTL')
eqtl = sub %>% filter(method == 'eQTL')
cur_mix = gen_fdr_power_curve(true_genes = true_eqtls, gene = mixqtl$eqtl, score = -log(mixqtl$pval), method = 'gt', cutoff = c(0, 1:20/10, 10:15/5, 4:50, Inf))
cur_eqtl = gen_fdr_power_curve(true_genes = true_eqtls, gene = eqtl$eqtl, score = -log(eqtl$pval), method = 'gt', cutoff = c(0, 1:20/10, 10:15/5, 4:50, Inf))
e = rbind(cur_mix %>% mutate(method = 'mixQTL') %>% filter(recall != 0) , cur_eqtl %>% mutate(method = 'eQTL') %>% filter(recall != 0))
pr_c[[length(pr_c) + 1]] = e %>% mutate(expression_level = i)
}
pr_c = do.call(rbind, pr_c)
pr_c$expression_level = factor(pr_c$expression_level, levels = c('high', 'middle', 'low'))
p2 = pr_c %>% ggplot() + geom_path(aes(x = recall, y = precision, color = method), size = 2, alpha = .8) + theme(aspect.ratio = 1) + facet_wrap(~expression_level)
p2 = p2 + th2
p2 = p2 + xlab('Recall') + ylab('Precision') + theme(legend.position = c(0.45, 0.2), legend.title = element_blank(), legend.text = element_text(size = 12)) + scale_color_manual(values = cbPalette)
p2
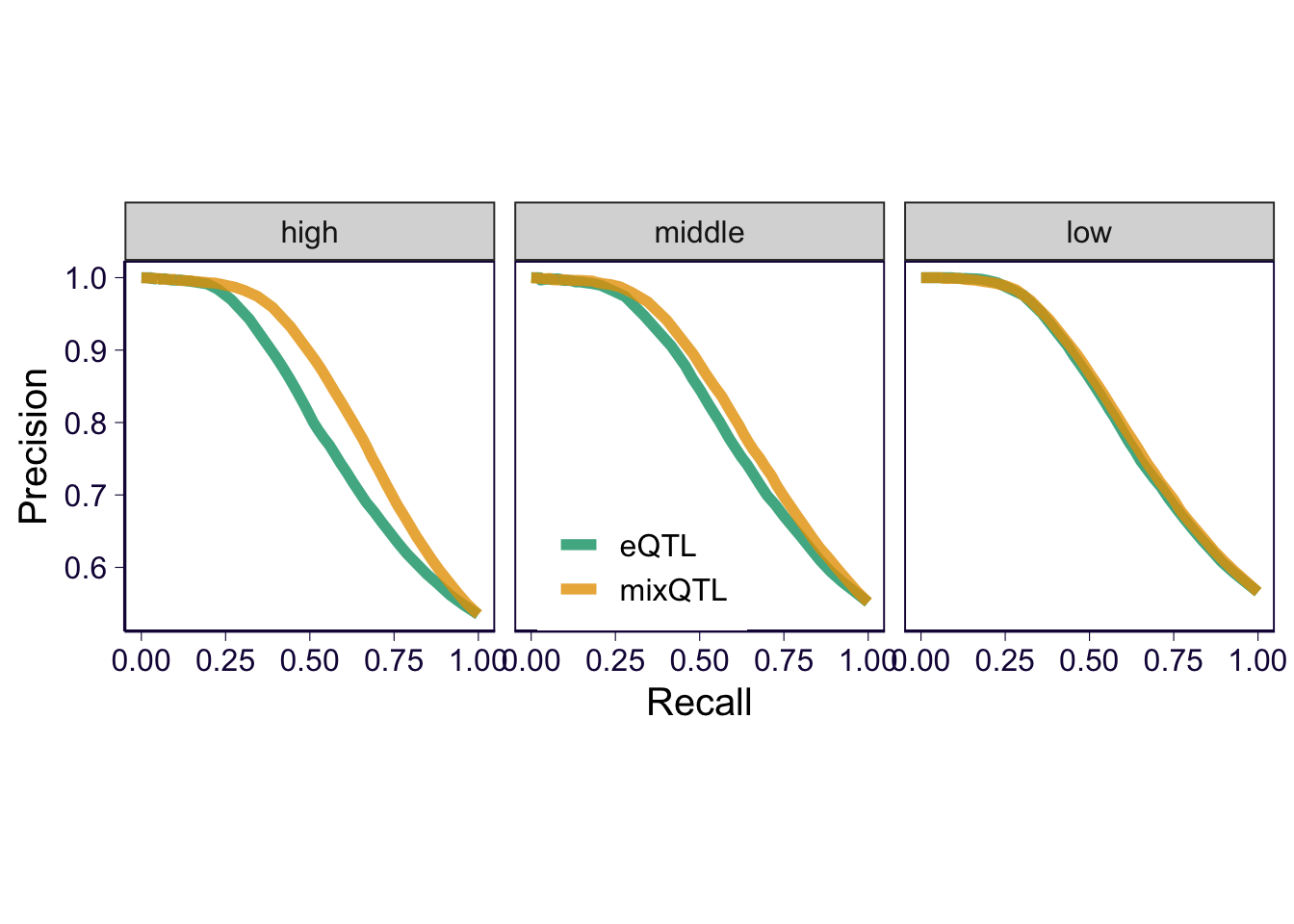
Save ROC/PR curves by expression level
ggsave('../output/gtex-v8-pipeline-eqtl-matrix-with-gene-qc-eqtlgen-roc-stratified.png', p, width = 10, height = 4)
ggsave('../output/gtex-v8-pipeline-eqtl-matrix-with-gene-qc-eqtlgen-pr-stratified.png', p2, width = 10, height = 4)